
🧬A Basic Biotech Primer
Part 1 of a First Principles Series - Decoding What Literally Defines Life


The $6 Billion Question
In 1999, the entire biopharmaceutical industry posted sales of roughly $6 billion. Half of that—$3 billion—came from just two proteins made by a single company: Amgen.
Think about that for a moment. Epogen, a protein that stimulates red blood cell production, and Neupogen, which revs up immune system cells in cancer patients, generated more revenue than thousands of other biotech companies combined. These weren’t complex molecules synthesized through years of chemical wizardry. They were copies—precise replicas of proteins your body already makes, manufactured by bacterial cells that had been taught a new trick.
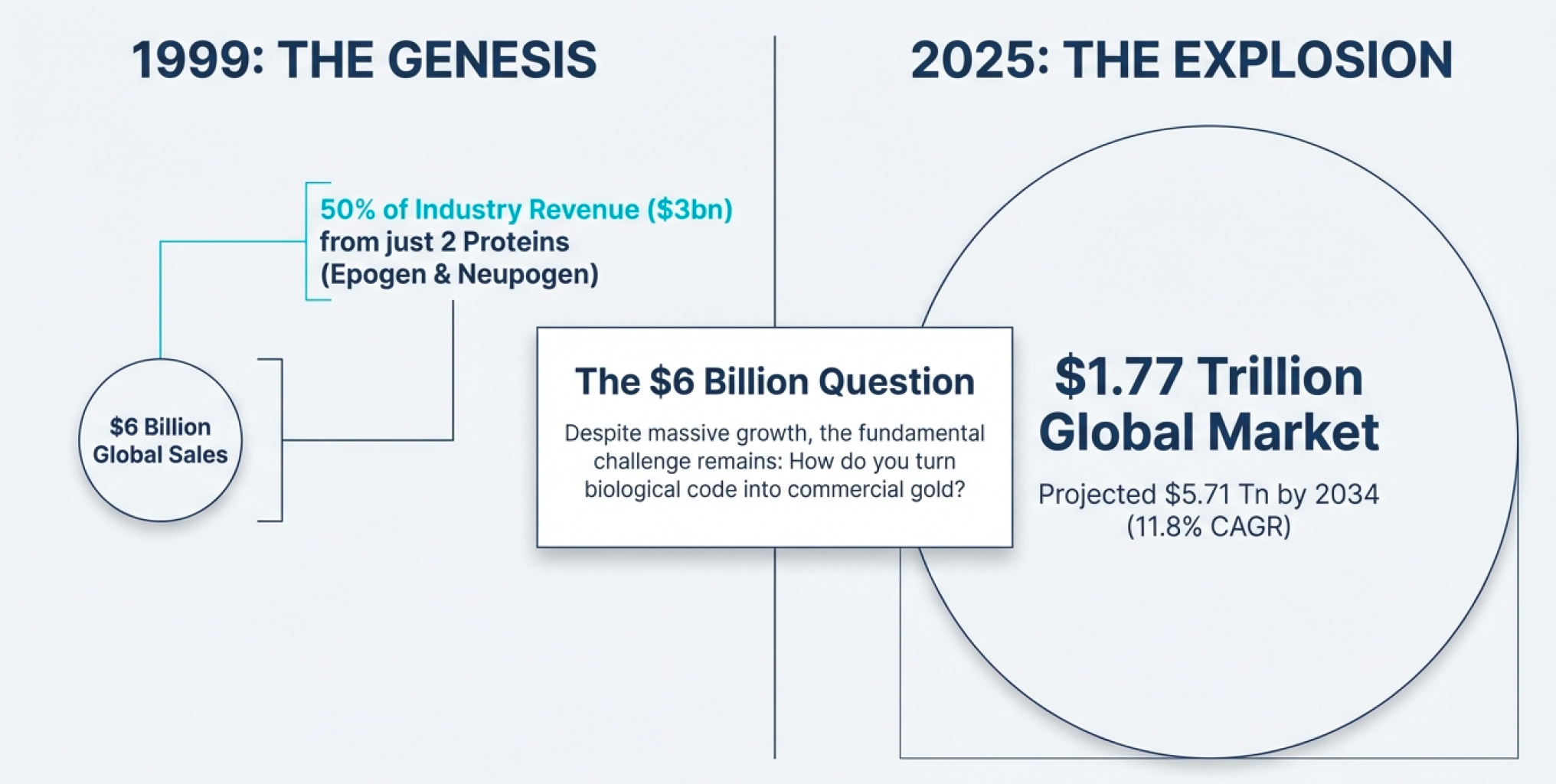
Fast forward to 2025, and the global biotechnology market stands at approximately $1.77 trillion, projected to hit $5.71 trillion by 2034. That’s a compound annual growth rate of 11.8%. North America alone accounts for $706 billion of this market. Yet the fundamental question remains unchanged: How do you turn biological code into commercial gold?
This isn’t a story about predicting the next miracle drug or timing the biotech cycle. It’s about understanding the infrastructure—the machinery, economics, and competitive dynamics that separate the companies manufacturing hope from those actually manufacturing cures. Because in biotech, unlike in most industries, the small players can legitimately compete with giants. You don’t see nameless automobile startups taking on GM, but you do see tiny biotech firms with breakthrough platforms threatening established pharmaceutical empires.
The question is: Why?
Part I: The Fundamental Problem — Why Your Body is Harder to Hack Than Your iPhone
The Protein Paradox
Here’s the dirty secret about modern medicine: we’ve been flying blind for most of human history. Imagine trying to fix a car when you can’t open the hood, can’t identify the parts, and the repair manual is written in a language that uses only four letters repeated three billion times. That’s biology.

The human body contains roughly 100,000 different types of proteins strung together across our chromosomes. Each protein is a gene expressed—a section of your DNA blueprint translated into a working molecular machine. Some proteins are structural (your bones, skin, hair). Others are communication systems (hormones, receptors). But the most fascinating are enzymes—proteins that catalyze every chemical reaction in your body.


When something goes wrong—cancer, diabetes, Alzheimer’s—it’s usually because a specific protein is malfunctioning. Either it’s doing something it shouldn’t, or it’s not doing something it should. The logical solution? Fix the protein, fix the problem.
Except there’s a catch: extracting enough protein to study it, let alone use it as a treatment, is extraordinarily difficult.
This is the original infrastructure problem in biotech. Before you can develop a cure for cancer, you need to get the proteins involved in cancer. You need sufficient quantities to understand how they work, test potential treatments, and eventually manufacture therapies at scale. For decades, this was done through painstaking extraction—grinding up tissues, filtering, purifying. It was slow, expensive, and yielded tiny amounts.
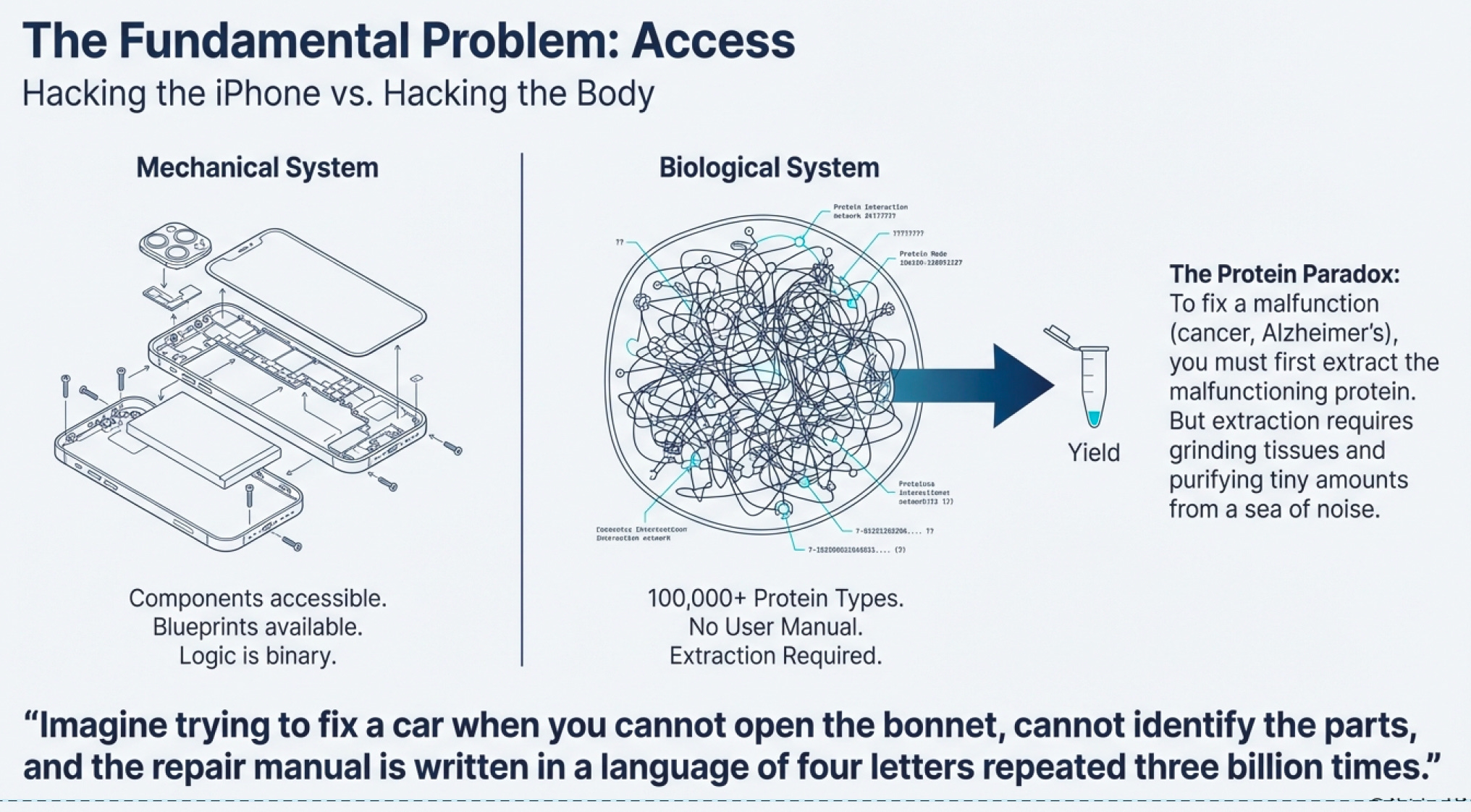
This scarcity is why, as noted in the biotech primer documents, there isn’t enough protein data in the market. Companies fail not because their science is wrong, but because they can’t overcome the fundamental infrastructure constraint: access to proteins.
The First Revolution: Teaching Bacteria to Build Human Parts
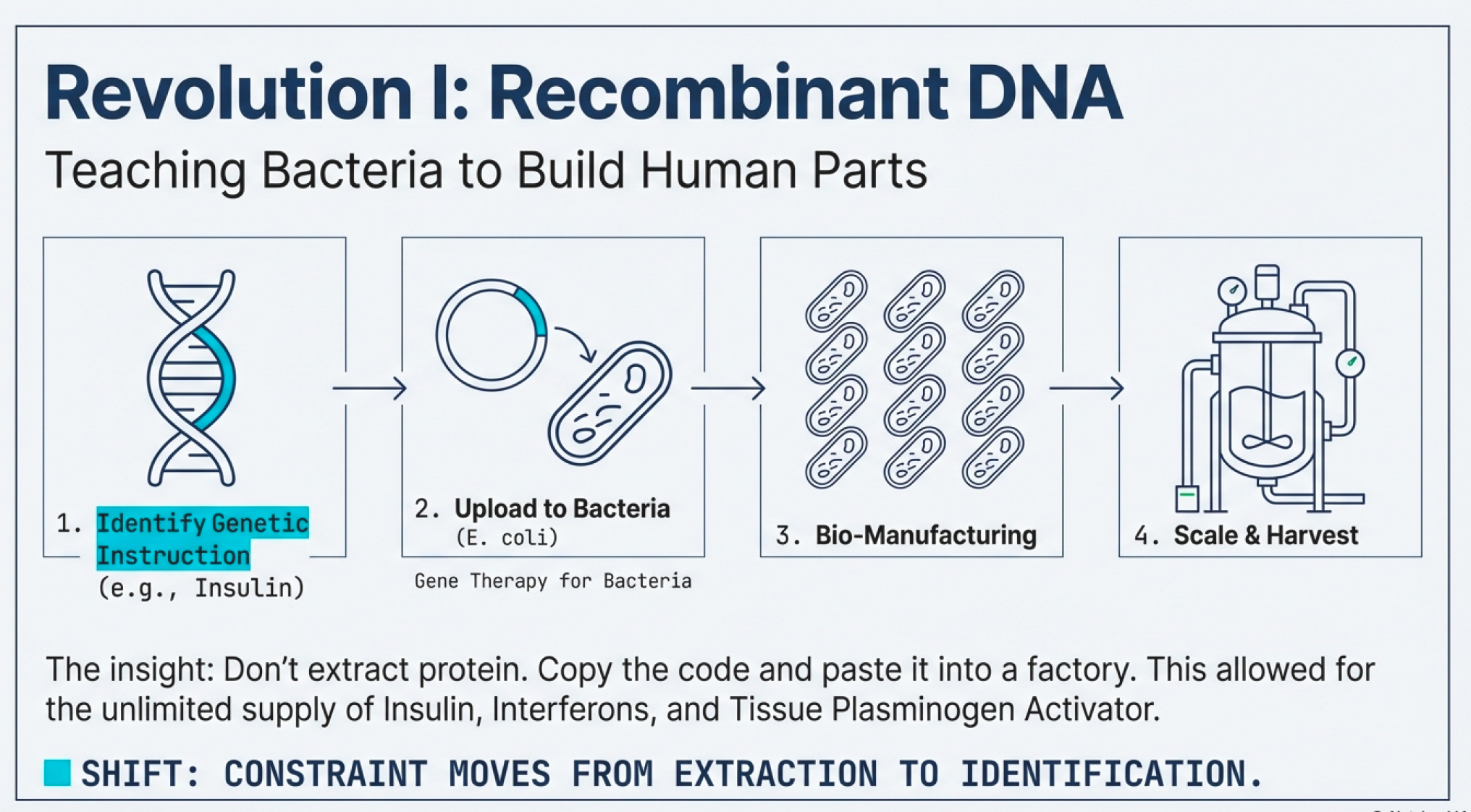
Then came recombinant DNA technology—arguably the most important infrastructure innovation in modern biotech.
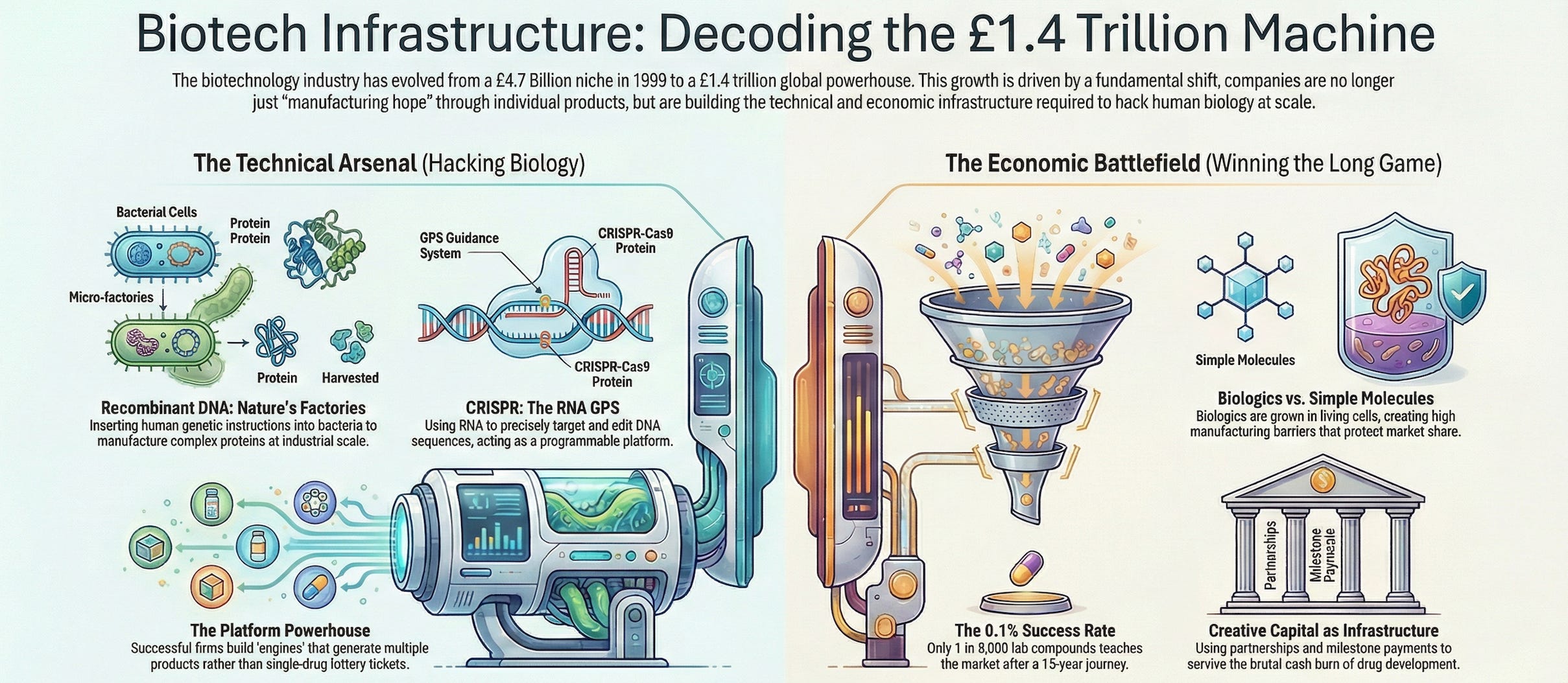
The insight was brilliant in its simplicity: instead of extracting proteins from human tissues, why not copy the genetic instructions for making those proteins and paste them into bacteria? Bacterial cells are nature’s factories. They reproduce rapidly, follow genetic instructions faithfully, and can be grown in massive vats. If you could teach E. coli to make human insulin, you’d have an unlimited supply.

This is not gene editing (a common misconception). Recombinant DNA is gene therapy—delivering engineered DNA into cells to counteract faulty genes. It doesn’t modify the patient’s DNA; it adds new instructions. Think of it as uploading new software rather than rewriting the operating system.
The commercial impact was immediate. Drugs like:
Interferons (slow cancer cell growth by improving the body’s natural response)
Tissue plasminogen activator (breaks up blood clots in heart attack victims)
Erythropoietin (stimulates red blood cell production)
Glucocerebrosidase (corrects genetic defects causing Gaucher’s disease)
All became manufacturable at scale. Suddenly, the infrastructure constraint wasn’t “can we extract enough protein?” but “can we identify the right genetic code and get bacteria to read it correctly?”
Gravity, Space Stations, and the Economics of Protein Crystal Formation
Here’s a delightful detour that illustrates how fundamental these infrastructure challenges are: gravity influences tissue culture, protein crystal formation, and nerve regeneration.
Why does this matter? Because to understand a protein’s structure—which determines its function—scientists often need to crystallize it. The quality of those crystals affects how well you can map the protein’s 3D structure using X-ray crystallography. In microgravity environments (like the International Space Station), proteins sometimes form better crystals because gravitational forces aren’t distorting the delicate formation process.
This is the level of infrastructure thinking required in biotech. You’re not just optimizing manufacturing efficiency or distribution networks. You’re wrestling with physics and chemistry at a molecular level. The infrastructure isn’t just factories and supply chains—it’s the fundamental scientific techniques that make biotech possible at all.
Part II: The Economic Battlefield — Where Small Players Slay Giants
The 0.1% Success Rate Problem
Let’s talk about the economics that make biotech investing simultaneously terrifying and thrilling.
The drug development pipeline looks like this:
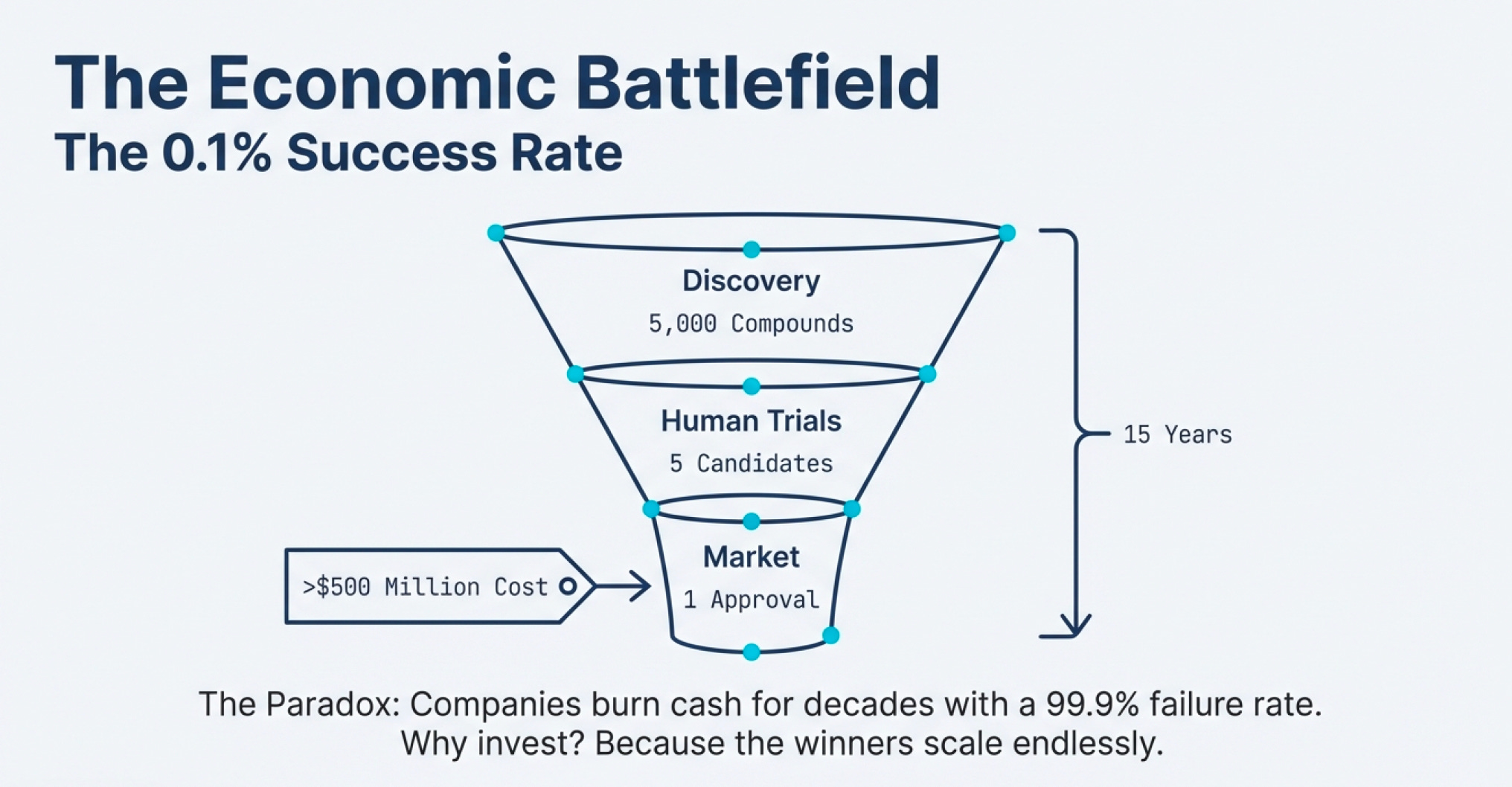
Discovery & Preclinical: For every 5,000 compounds that show promise in the lab and in animal testing, only 5 make it to human trials.
Clinical Trials (Phase 1, 2, 3): Of those 5, only 1 eventually reaches the marketplace.
FDA Approval: The entire process takes an average of 15 years and costs around $500 million (in 1999 dollars; today it’s significantly higher).
That’s a 0.02% success rate from initial discovery to market—or a 0.1% success rate if you count only compounds that make it past preclinical testing.

Think about what this means for business economics. If you’re a traditional investor evaluating companies by their near-term cash flows, biotech looks insane. Companies burn money for years—sometimes decades—before generating revenue. Most products fail. The ones that succeed might take 15 years to pay off.
Yet the industry works. Why?
The Platform vs. Product Paradox
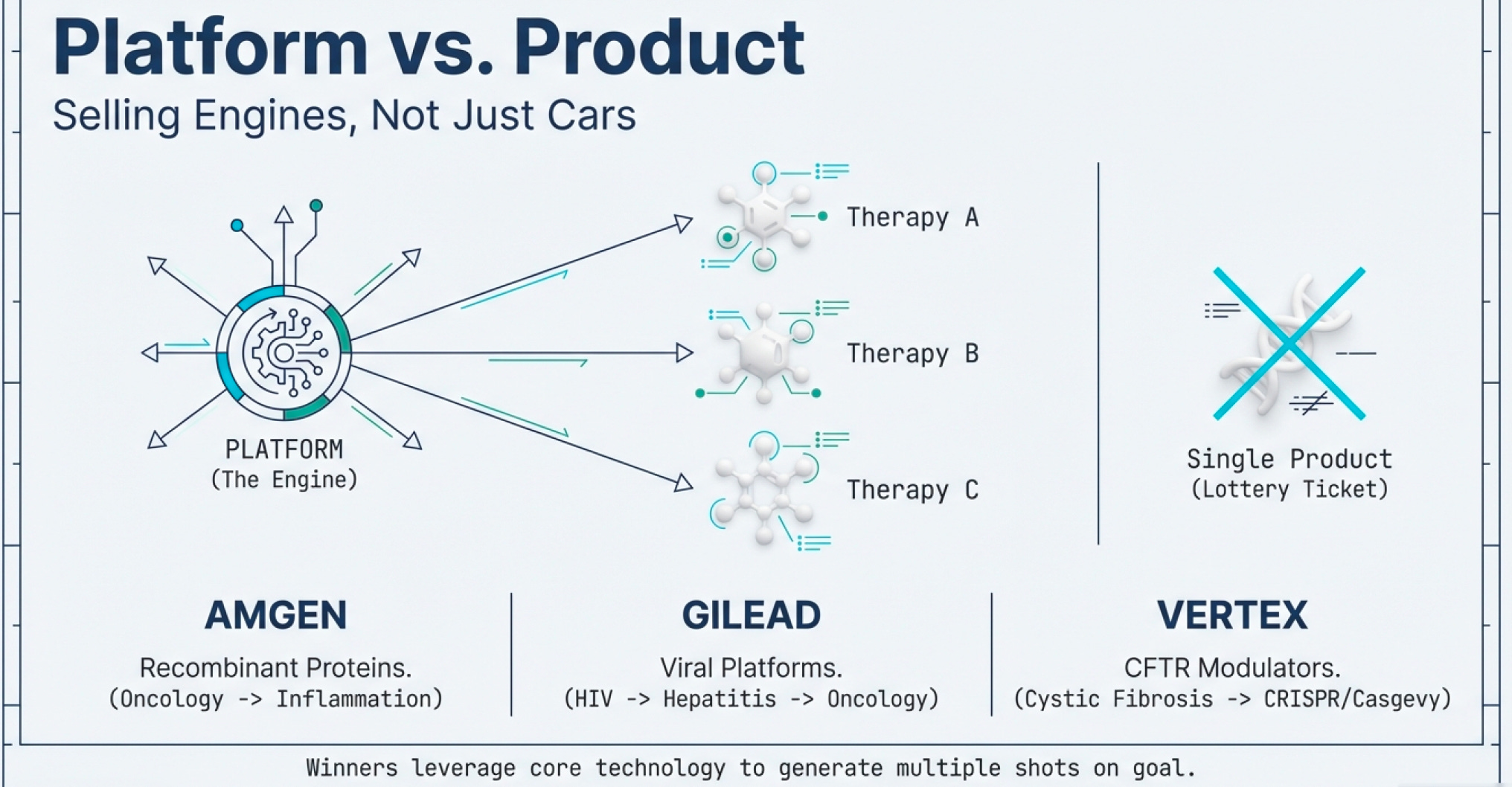
The key insight: great biotech companies don’t sell products; they sell platforms.
Recombinant DNA wasn’t just insulin; it was a manufacturing method applicable to hundreds of proteins. CRISPR isn’t just a sickle cell treatment; it’s a genome editing platform applicable to thousands of genetic diseases.
This is why, despite the $1.77 trillion global market in 2025, small companies can compete. They’re not competing on manufacturing scale or distribution networks (though those matter eventually). They’re competing on technological innovation infrastructure—on having a better mousetrap for solving the fundamental protein problem. Historically, big Pharmas don’t seem to fare much better than small Biotech companies making waves.
Consider the current competitive landscape among pure-play biotech leaders:
Amgen (market cap >$100 billion): Specializes in biologics including monoclonal antibodies and recombinant proteins for oncology, nephrology, and inflammation. In September 2025, Amgen announced a $600 million expansion of its Thousand Oaks headquarters—a new research center bringing together researchers, engineers, and scientists for “the next era of scientific discovery.” The company employs 5,500 people at its Ventura County campus alone.
Gilead Sciences (market cap >$100 billion): Originally focused on antivirals (including groundbreaking HIV treatments), Gilead has expanded aggressively into oncology through acquisitions like Kite Pharma (CAR-T therapies) and Immunomedics (Trodelvy for cancer). In September 2025, Gilead broke ground on an $847 million pharmaceutical development and manufacturing center at its Foster City campus, expected to support 2,500 jobs when completed in 2029.
Vertex Pharmaceuticals (market cap >$100 billion): Pioneered treatments for cystic fibrosis and co-developed Casgevy, the first FDA-approved CRISPR therapy (approved December 2023 for sickle cell disease). Vertex records 100% of Casgevy revenues while sharing net profits 60/40 with CRISPR Therapeutics.
Regeneron Pharmaceuticals (market cap >$100 billion): Known for innovative biologics and antibody treatments. Competes directly with Amgen in several therapeutic areas.
Genentech (owned by Roche): A biotechnology pioneer, remains formidable in oncology with a revenue ranking 15th among top competitors (averaging $39.2 billion), though with 13,539 employees it ranks 19th in workforce size among top 10 competitors (averaging 54,667 employees).
Notice the pattern? These companies aren’t winning primarily through scale economies in manufacturing and specializing in 1 winning product. They’re winning through platform capabilities—the ability to generate multiple products from core technology.
The Tool Company Trap
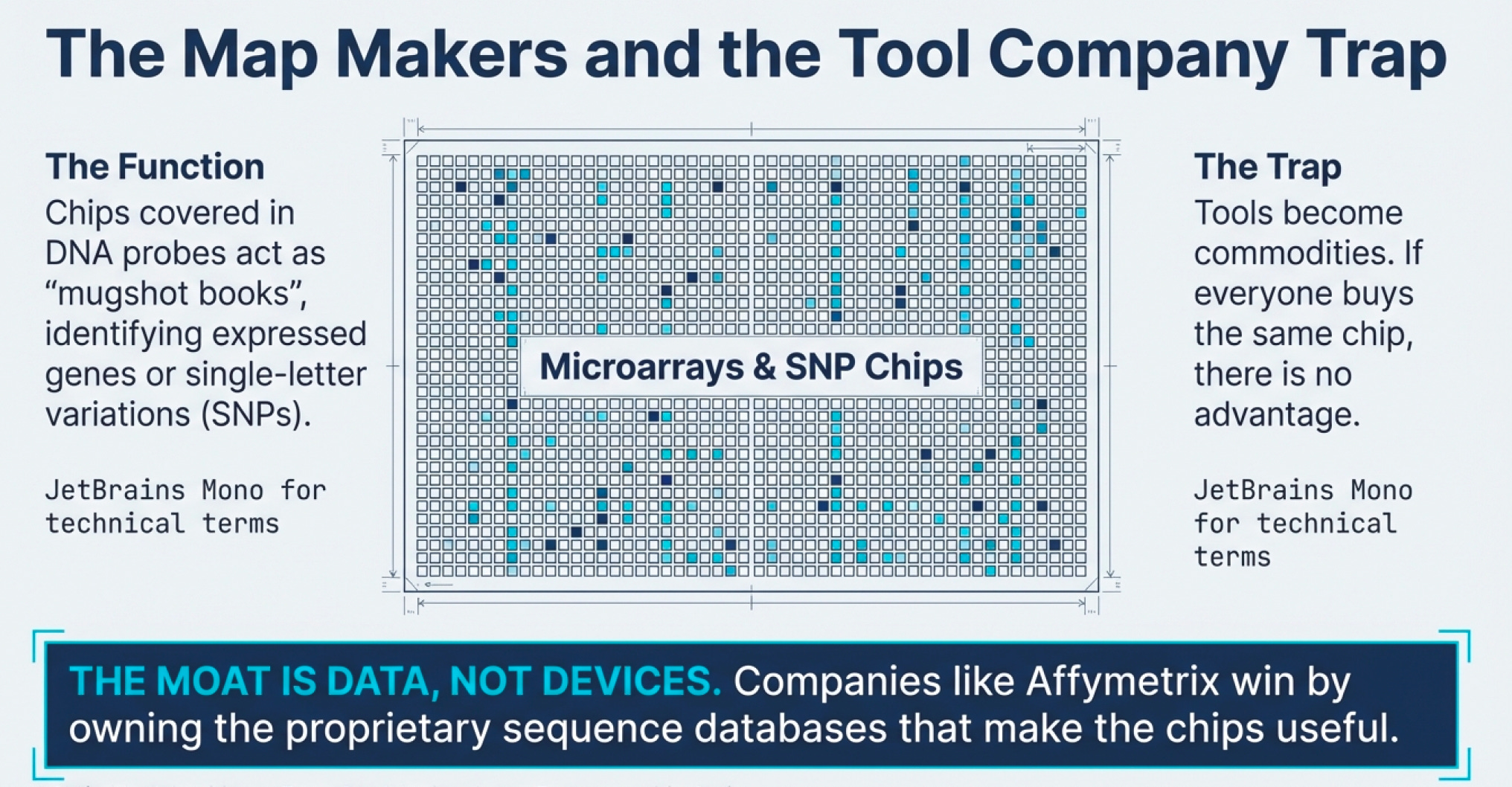
Here’s where many investors go wrong: they fall in love with “tool companies”—firms selling picks and shovels to the biotech gold rush.
The logic seems sound: if biotech companies need microarrays, chromatography equipment, or DNA sequencing services, why not invest in the companies providing those tools? You’re insulated from the drug development risk!
The problem? Tools become commodities.
When a tool company licenses its technology to many different drug developers, everyone possesses the same capability. There’s no sustainable competitive advantage. The tool might be cutting-edge today, but in biotech, today’s revolution is tomorrow’s baseline expectation. Unless the tool company has proprietary sequences (like Affymetrix’s agreement with Human Genomics Sciences for access to proprietary database sequences) or some other sustainable moat, it’s fighting commodity economics.
Creative Financing as Infrastructure
In biotech, capital structure is infrastructure.
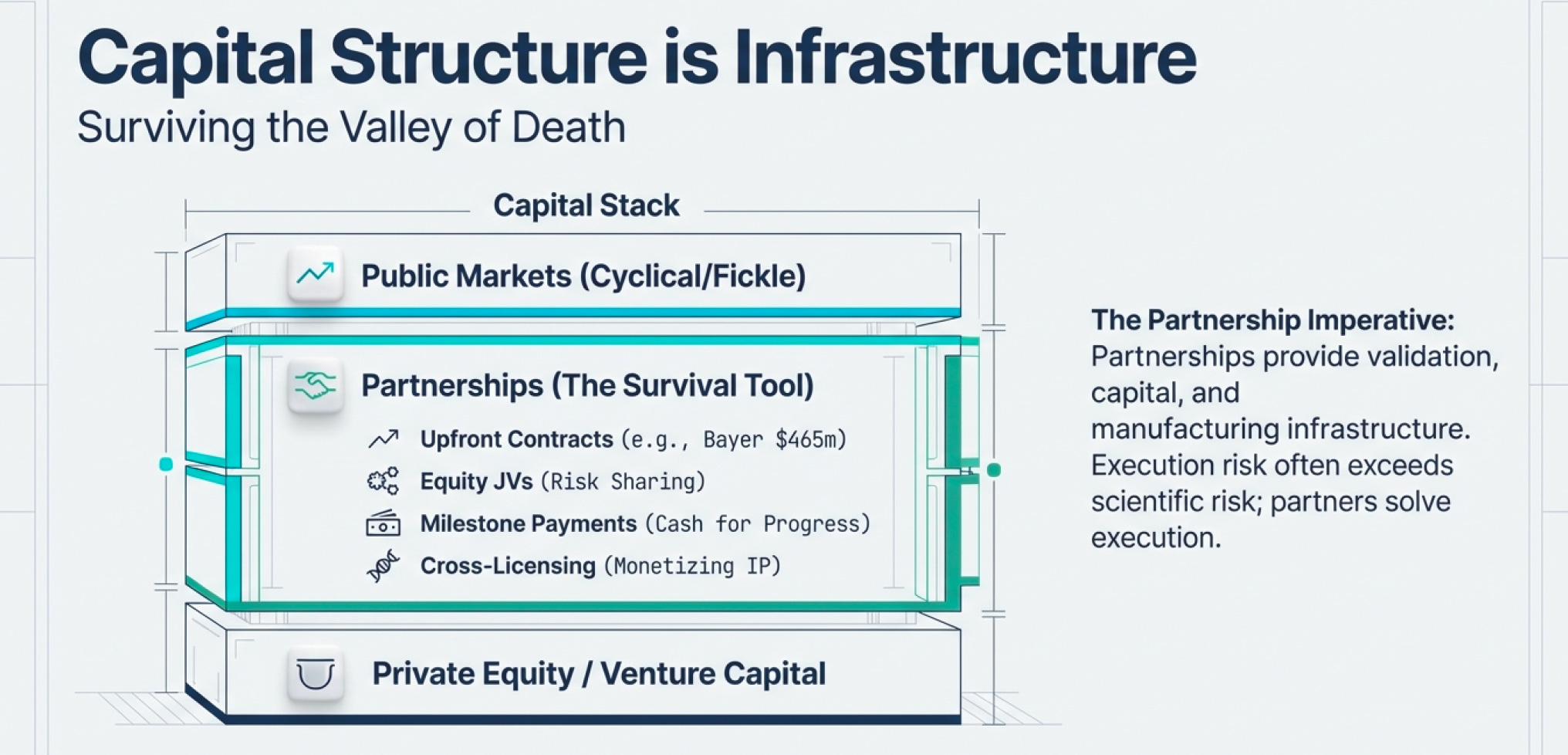
Because the sector burns through cash (due to R&D) and most public market investors lack technical knowledge to evaluate science, biotech companies must be creative about financing. The ability to access capital when public markets are closed isn’t a nice-to-have—it’s existential.
Consider Millennium Pharmaceuticals’ financing playbook (a case study in biotech capital strategy):
Large upfront target-discovery contracts: Bayer (1997) paid $465 million for multiyear access to Millennium’s target discovery capabilities across oncology, cardiovascular, osteoporosis, and pain. This gave Millennium predictable cash to scale operations.
Equity-backed joint ventures: Eli Lilly took an equity stake and co-funded Millennium BioTherapeutics in the mid-1990s. This shared risk while giving both parties upside on discoveries.
Milestone payments: Lilly paid Millennium as they delivered validated targets—essentially converting R&D progress into near-term cash that could fund subsequent work.
Cross-industry platform licensing: Monsanto paid $218 million over five years for Millennium to apply its genomics platform to crop traits. This generated revenue outside pharma cycles entirely.
Co-development profit-sharing: Aventis partnered on inflammation programs ($450 million over five years) with equal U.S. profit sharing. This split late-stage costs while aligning incentives.
Out-licensing: Distribution deals with larger companies (like Schering/Berlex for manufacturing and marketing) freed capital by avoiding expensive infrastructure buildout.
The result? Millennium could fund ambitious R&D through partnerships, maintaining optionality without diluting early shareholders excessively.
Other creative structures include:
Tracking stocks: Allow companies to retain IP assets while issuing tax-free incentives to partners/investors for specific business units
Private placements, PIPEs, and Regulation S deals: Selling public shares at a discount to sophisticated investors (or overseas investors for Reg S)
Convertible preferred stock: Often with resets and escalating conversion discounts as milestones are hit
Revenue-based transactions: Trading uncertain future royalty rights for immediate cash (like Xoma’s deal with Genentech or Cephalon’s with Provigil)
These aren’t financial engineering tricks—they’re infrastructure for surviving the 15-year development timeline.
The Partnership Imperative
Here’s the business reality: biotech companies partner aggressively because (1) they need to survive the sector’s brutal burn rate, and (2) they need to validate demand for public market investors who can’t evaluate the science.
Hoffmann La-Roche accumulated over 120 biotech alliances between 1988 and the late 1990s. Why? Because partnerships provide:
Validation: A major pharma company betting on your science signals credibility
Capital: Upfront payments, milestone payments, and profit-sharing reduce cash burn
Infrastructure: Access to manufacturing, regulatory expertise, and distribution networks
Optionality: Freedom to pursue multiple programs without over-committing to any single bet
The most successful biotech companies aren’t precious about their discoveries. They understand that execution risk (can we actually manufacture and sell this?) often exceeds scientific risk (will this work?). Partnering with organizations that have proven infrastructure reduces execution risks (and at many times, provide liquidity/ working capital).
Part III: The Technical Arsenal — Weapons for the Molecular War
CRISPR: When Science Fiction Becomes Infrastructure
Let’s talk about arguably the most important biotech infrastructure innovation since recombinant DNA: CRISPR.
CRISPR started as a curiosity—an odd bacterial immune system. Bacteria capture snippets of viral DNA and insert them into their own genome in repeating patterns (hence “Clustered Regularly Interspaced Short Palindromic Repeats”). These CRISPR arrays act as “mugshots” of dangerous viruses. When the same virus attacks again, the bacteria produce RNA segments from the CRISPR array that recognize and attach to the virus’s DNA. Then an enzyme called Cas9 cuts the viral DNA apart, disabling the invader.

The insight: this could be repurposed for precise genome editing in any organism.
Here’s how it works:
Design a guide RNA (crRNA): A short sequence that matches the DNA target you want to edit
Add tracrRNA: A “handle” that helps Cas9 grip the crRNA firmly (later combined into single-guide RNA/sgRNA)
Load the system: The guide RNA + Cas9 protein forms a complex
Search: The complex scans DNA looking for PAM sequences (a specific 3-letter code like NGG)
Cut: When the guide RNA matches the target perfectly, Cas9 cuts both DNA strands
Repair: The cell’s natural repair machinery either:
Non-homologous end-joining: Stitches ends together (often sloppy, causing deletions/insertions)
Homology-directed repair: Uses a provided template to insert specific sequences

The infrastructure advantage over previous gene-editing tools (like TALENs)? CRISPR uses RNA as its GPS instead of protein. This means you don’t have to construct a new protein every time you want to target a different sequence—you just change the RNA guide. It’s dramatically faster and cheaper.

But there was a challenge: getting Cas9 into the human cell nucleus (since it was not previously used in human cells). Two solutions:
Codon optimization: Ensuring the bacterial Cas9 gene works efficiently in human cells
Nuclear localization signals: Tagging Cas9 with a molecular “zip code” for active transport into the nucleus
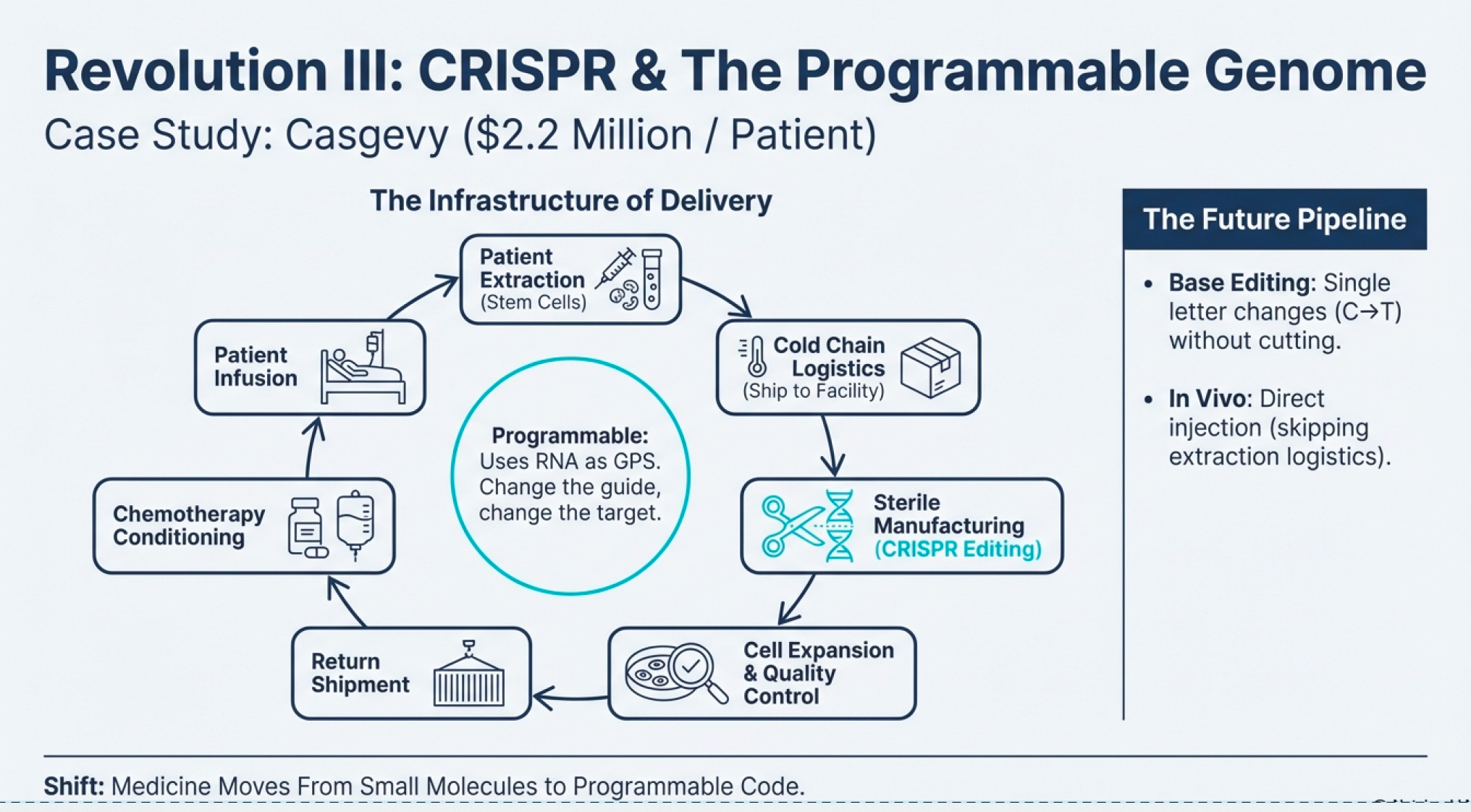
The First Functional Cure: Casgevy and the $2.2 Million Question
On December 8, 2023, the FDA approved Casgevy (exagamglogene autotemcel)—the first CRISPR/Cas9 genome-edited therapy for human use. It treats sickle cell disease in patients 12 years and older with recurrent vaso-occlusive crises.
The disease mechanism: A single-letter mutation in the β-globin gene causes hemoglobin to polymerize abnormally, forming long fibers that contort red blood cells into sickle shapes. These sickled cells clump together, blocking blood vessels and causing excruciating pain, organ damage, and early death (typically by age 50).

The CRISPR solution: Instead of directly fixing the mutation (which is technically challenging), Casgevy targets the BCL11A gene—a regulator that normally shuts off fetal hemoglobin (HbF) production after birth. By disabling BCL11A in blood stem cells, the cells continue producing HbF throughout adulthood. HbF carries oxygen efficiently and prevents sickling.
The clinical results are stunning:
Of 31 patients treated in the pivotal trial, 29 had no vaso-occlusive crises for at least one year after treatment
Patients are followed for only 18-24 months so far, but early indications suggest durability
As of spring 2025, Casgevy has been approved in the US, UK, EU, Switzerland, Canada, Bahrain, Saudi Arabia, and UAE. There are 50 active treatment centers globally, and over 50 patients have begun treatment.
But here’s the infrastructure problem: Casgevy costs $2.2 million per patient.
This isn’t price gouging—it’s the economics of the infrastructure required:
Extract the patient’s hematopoietic stem cells via bone marrow harvest
Ship to a specialized manufacturing facility
Perform CRISPR editing on the cells in a sterile environment
Expand the edited cell population to therapeutic quantities
Quality-test extensively to ensure successful editing and no off-target effects
Ship back to the treatment center
Pre-condition the patient with chemotherapy (to make room in bone marrow for engraftment)
Infuse the edited cells
Monitor intensively for months as engraftment occurs
The entire process takes months and requires highly specialized laboratories. Only about 50 facilities worldwide can perform it. CRISPR Therapeutics and Vertex are working on reimbursement models with U.S. state Medicaid programs and the UK’s National Health System based on outcomes—if the treatment works (eliminates pain crises), payers reimburse; if not, they don’t.
This is biotech infrastructure thinking: the scientific breakthrough is only half the battle. The delivery system matters just as much.
Beyond Casgevy: The CRISPR Pipeline
CRISPR isn’t stopping at sickle cell. The clinical pipeline includes (variations of CRISPR):
Base Editing (Beam Therapeutics): Instead of cutting DNA, base editors change single letters (A→G, C→T) without creating double-strand breaks. In January 2024, Beam dosed the first patient in a Phase 1/2 trial for severe sickle cell disease using base editing. Early data from 7 patients (presented December 2024) showed >60% HbF induction and <40% HbS reduction with resolution of anemia in all seven.
Prime Editing: Allows insertion of longer sequences (up to 80 letters) with only a tiny nick in DNA rather than a double-strand break. Think of base editing as a pencil and prime editing as a word processor.
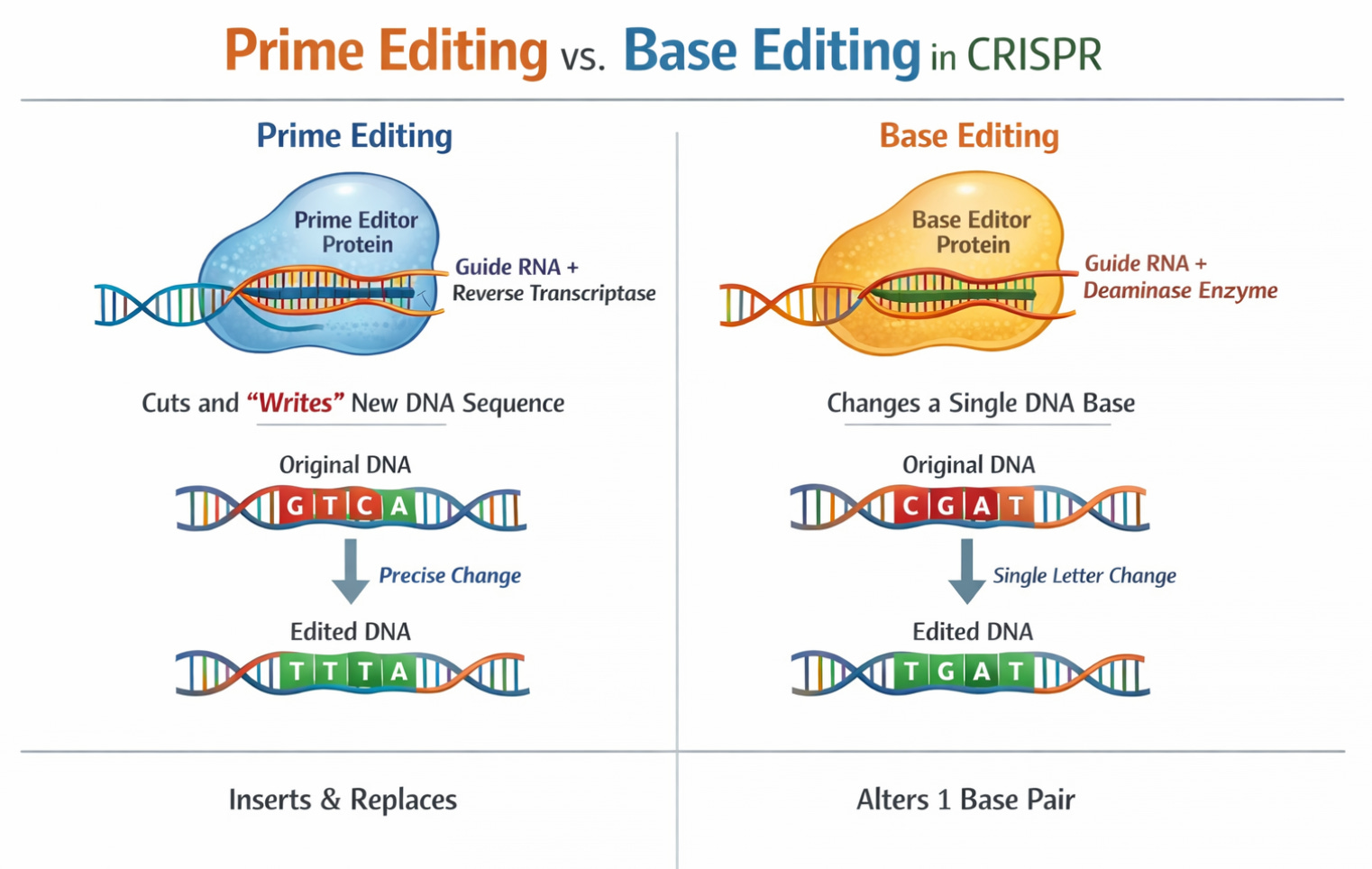
In Vivo CRISPR: All current approved therapies are ex vivo (cells extracted, edited outside the body, returned). But delivering CRISPR directly into the body opens possibilities for treating diseases where cell extraction isn’t feasible. Editas Medicine’s EDIT-101 trial (treating Leber congenital amurosis, a genetic blindness) involves direct injection into the retina. Early results (published May 2024 in NEJM) show meaningful vision improvements in 6 of 14 participants.
Monoclonal Antibodies: The Other Revolution
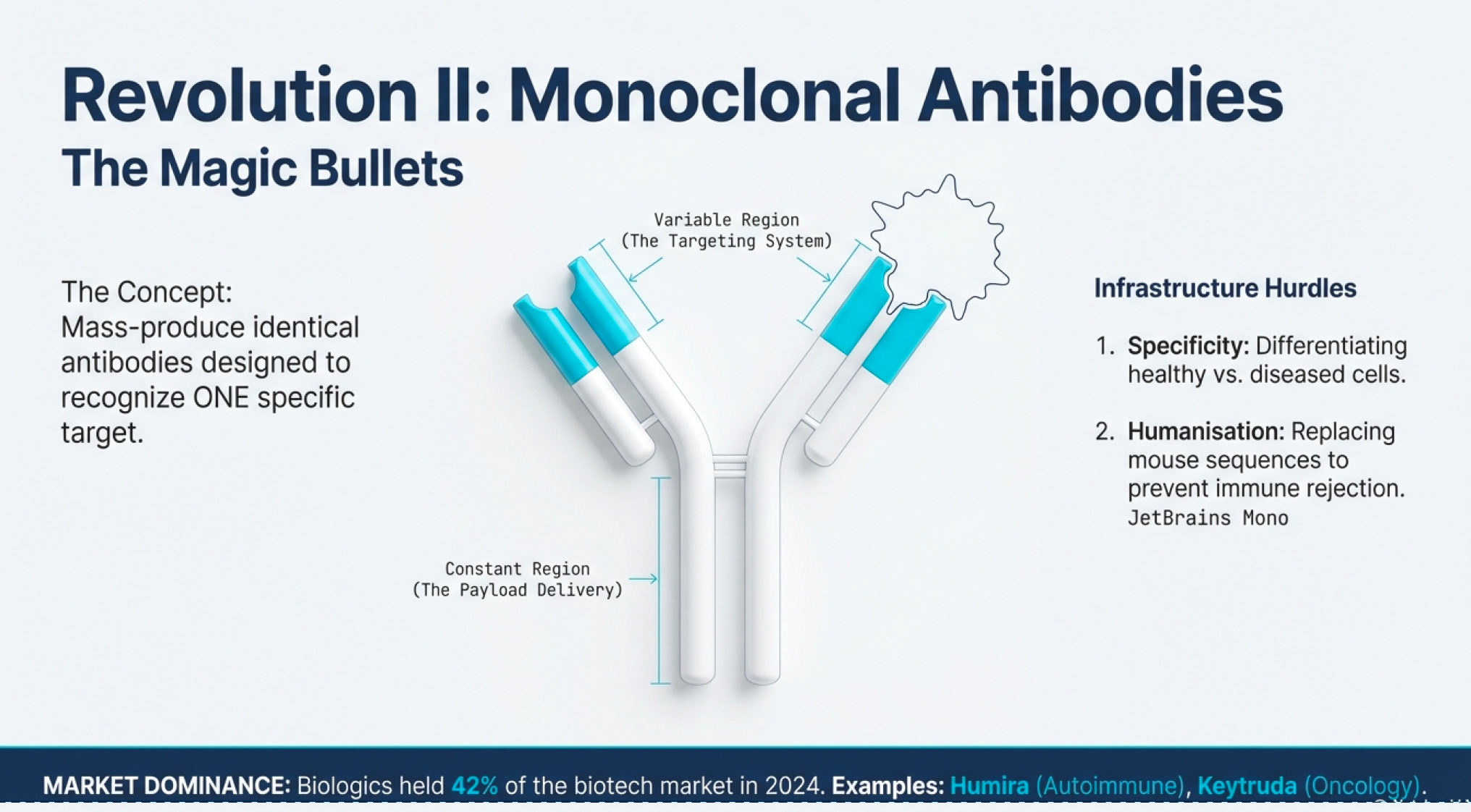
CRISPR and recombinant DNA are the flashy innovations, but monoclonal antibodies might be biotech’s most commercially successful infrastructure advance.
Antibodies are proteins your immune system produces to recognize foreign invaders (viruses, bacteria, cancer cells). They bind to specific targets and signal the immune system to attack. The problem with natural antibodies: your body produces millions of different antibodies, each recognizing different targets. Isolating a specific antibody for a specific target was historically impossible.
Enter monoclonal antibody technology: a method for producing huge quantities of identical antibodies targeting a specific molecule. This became known as “magic bullets”—you could design an antibody that recognizes only cancer cells, attach a toxic payload, inject it into the bloodstream, and it would deliver chemotherapy directly to tumors while sparing healthy tissue.
Two major infrastructure hurdles:
Specificity: Finding antibodies that differentiate between healthy cells and diseased cells. Cancer biology is complex—tumor proteins are often just mutated versions of normal proteins.
Immune Recognition: The immune system tends to recognize therapeutic antibodies as foreign (especially if derived from mice) and eliminate them before they work. Solution: “humanize” antibodies by replacing mouse protein sequences with human equivalents.
The commercial applications exploded beyond cancer:
COVID-19: Monoclonal antibodies were used to prevent virus-receptor binding (like blocking the spike protein from attaching to ACE2 receptors)
Autoimmune diseases: Antibodies targeting inflammatory cytokines (like TNF-α) revolutionized treatment of rheumatoid arthritis and Crohn’s disease
Diagnostics: Antibodies that bind to specific disease markers enable rapid testing
The global biologics market (which includes monoclonal antibodies) held a 42% share of the biotechnology market in 2024 and is projected to grow at 13.9% CAGR through 2030. Biopharmacy applications—primarily monoclonal antibodies and recombinant proteins—dominate the biotech landscape.
The Diagnostic Infrastructure: Microarrays and SNP Chips
Here’s a less glamorous but critical piece of infrastructure: identification systems.
Before you can treat a disease, you need to diagnose it precisely. And increasingly, precision medicine requires understanding genetic variations.
Microarrays are essentially DNA hybridization platforms. They work on a simple principle: complementary DNA strands stick together (A pairs with T, G pairs with C). A microarray is a chip covered with thousands of known DNA sequences (called probes). When you wash a patient’s DNA (with a die e.g. green or red fluorescent) over the chip, it binds to matching probes. You can then identify which genes are present (as the fluorescent shows up), which are mutated, and how they’re expressed.

SNP chips (single nucleotide polymorphism chips) are a specialized microarray that detect specific genetic variations. SNPs are single-letter differences in DNA sequence (like having an A instead of T at position 1,284,556 of chromosome 7). These variations determine everything from eye color to Alzheimer’s risk to drug response.
The infrastructure components that matter:
Probe Libraries: How many different sequences can the chip detect? More probes = more comprehensive testing
Density: How many probes per square centimeter? Higher density = smaller samples needed
Speed: How quickly can you run the test? Faster = more patients screened
Accuracy: How many false positives/negatives? Higher accuracy = better clinical decisions
Price: Cost relative to clinical value
Companies like Affymetrix gained competitive advantages through proprietary sequence databases—essentially exclusive “mugshot books” of genetic variations that competitors couldn’t access. This is the biotech equivalent of having better maps than your competitors.
The diagnostic infrastructure enables pharmacogenomics—understanding how genetic differences affect drug response. One person might metabolize a drug rapidly (requiring higher doses), while another metabolizes it slowly (risking toxicity at normal doses). SNP chips can identify these variations before prescribing.
The COVID-19 Diagnosis Story: CRISPR-Cas12 and Cas13
The pandemic illustrated how biotech infrastructure innovations can be rapidly deployed for new threats.
Mammoth Biosciences discovered that Cas12, like Cas9, can find and cut specific DNA sequences. But unlike Cas9, which stops after making its cut, Cas12 goes into an “indiscriminate cutting frenzy,” chopping up any nearby single-stranded DNA. Mammoth combined this with fluorescent reporter molecules: when Cas12 found target COVID DNA, it would also chop up reporters, causing them to glow—a positive test signal.
Sherlock Biosciences discovered Cas13, which targets RNA instead of DNA (Cas9 & Cas12) and has the same frenzy-cutting behavior.
Since COVID-19’s genetic material is RNA, Cas13 seemed better suited. But there was a complication:
To amplify RNA for detection, you must first convert it to DNA (reverse transcription) e.g. instead of just testing 1 by 1 with 1 sample, by amplifying, you create multiples “clones” to test, which makes the test faster
After amplification, transcribe back to RNA
Then use Cas13 to detect the RNA
The Cas12 (targetting DNA) test could skip step 2-3, going directly from amplified DNA to detection. Both systems worked, but the discovery of Cas13 was significant because most human-threatening viruses have RNA genomes. This infrastructure—CRISPR-based diagnostic platforms—can be rapidly retooled for new pathogens.
Part IV: The Infrastructure Mindset - What Separates Winners from Losers
The Five Pillars of Great Biotech Companies
Based on decades of biotech evolution, successful companies consistently exhibit:
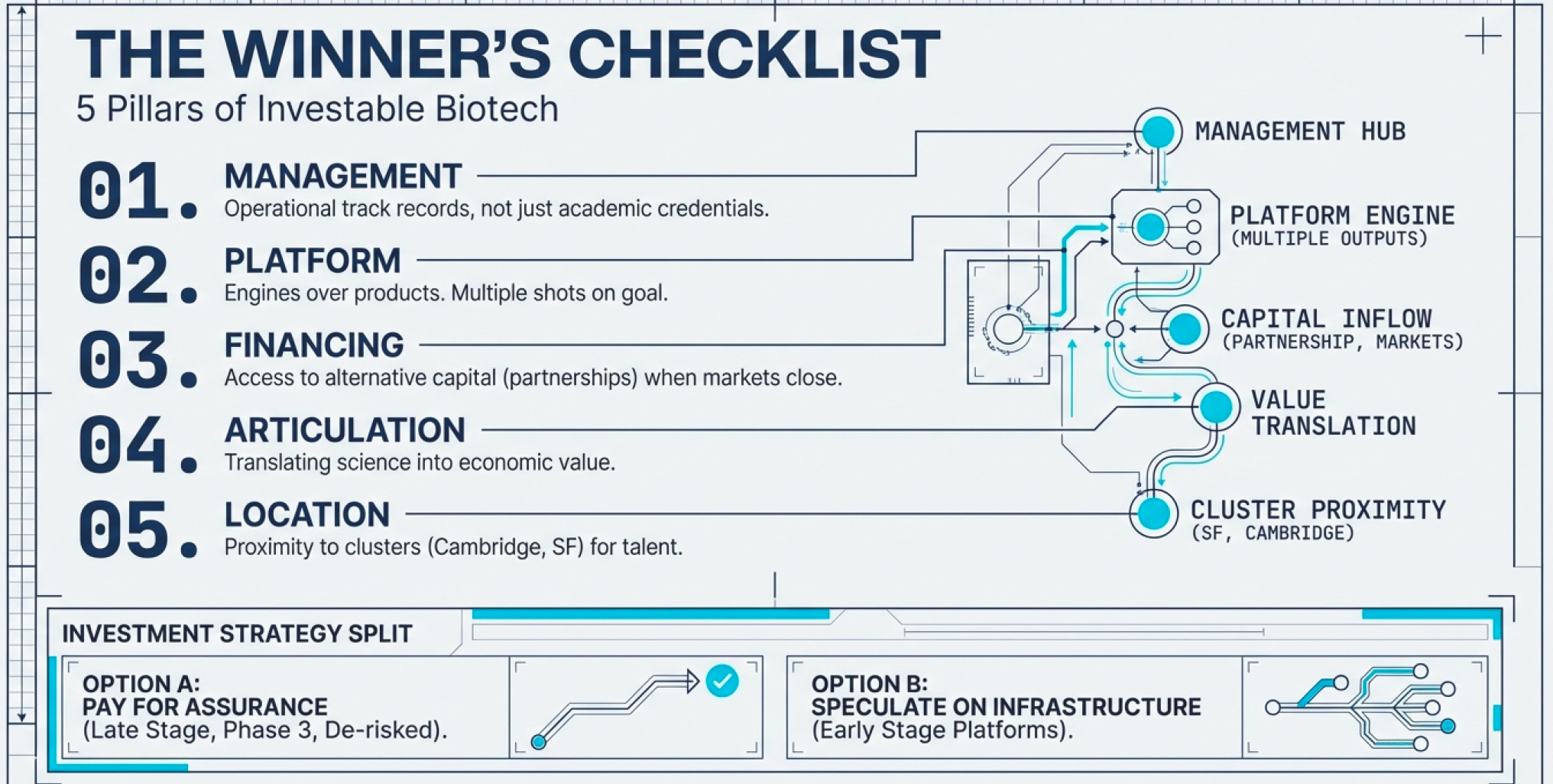
1. Excellent Management with Industry Experience Science brilliance isn’t enough. You need leaders who understand regulatory pathways, manufacturing scale-up, commercial partnerships, and financing strategy. The best biotech CEOs have operational track records, not just academic credentials.
2. Platform Technology (Not Just Products) Single-product companies are lottery tickets. Platform companies have engines for generating multiple product opportunities (remember that it takes an average of 15 years to clear a drug and the chances of that happening is 0.1%).
3. Access to Alternative Financing When public markets close (and they will—biotech is cyclical), you need backup capital sources: corporate partnerships, private placements, debt facilities, royalty monetization deals. Companies that depend solely on public equity raises die in downturns.
4. Ability to Articulate Business Opportunity Scientists often struggle to translate technical achievements into economic value propositions. Great biotech companies have translators—people who can explain to investors, regulators, and partners why this matters and how it makes money.
5. Strategic Location Biotech clusters exist for good reason. Being near top research universities (MIT, Stanford, UCSF, UCSD) provides access to talent and early-stage research. Being near manufacturing facilities and specialized service providers reduces friction. California dominates biotech (47% of North American market share in 2022) because the ecosystem infrastructure is unmatched.
The Clinical Development Strategy
Here’s where infrastructure thinking meets execution: clinical trial design is a competitive weapon.
Consider two approaches:
Broad indication first: Try to treat a large patient population (e.g., all cancer types). This maximizes potential market but increases regulatory complexity and trial costs. Failure risk is high because the biology varies across the broad population.
Narrow indication first, expand later: Target a small, well-defined patient population where the biological mechanism is clear (e.g., rheumatoid arthritis patients with a specific genetic marker). Get FDA approval for this niche. Generate real-world evidence. Then expand to related populations in follow-up studies.
The second strategy is almost always superior. It allows you to:
Move faster (smaller trials = faster enrollment)
Spend less (fewer patients = lower costs)
Prove concept before scaling (de-risk subsequent indications)
Generate revenue sooner (even a small market is better than no market)
Build clinical data that supports expansion
This is infrastructure thinking. The trial design IS the infrastructure for commercialization. Design it poorly, and even great science fails.
The Investment Implications: Two Strategies
For investors (and this is not investment advice, just structural observation), two coherent approaches exist:
1. Pay for Assurance: Stock up on companies with product candidates in late stages (Phase 3 or awaiting FDA approval). These companies have:
De-risked science (Phase 2 proved it works)
Clear regulatory path (FDA guidance on approval requirements)
Near-term revenue catalysts (approval within 12-24 months)
The tradeoff: you’re paying premium valuations. But Van Den Broek’s research found that even waiting until final FDA approval, you could still outperform the NASDAQ average. Why? Because most investors underestimate the commercial potential of approved drugs until revenue materialized.
2. Speculate on Cheap Small Caps: Look for platform companies with multiple shots on goal, great management, and access to alternative financing. These are lottery tickets, but with better odds than true wildcards because:
Platform reduces single-product risk
Good management reduces execution risk
Alternative financing reduces capital risk
The ideal scenario: cheap small cap with multiple Phase 2/3 candidates, strong partnerships, excellent management, and a bear market in biotech (forcing valuations down despite solid fundamentals).
What NOT to Do
Don’t chase tool companies without sustainable moats. If everyone has access to the same technology, it’s a commodity.
Don’t ignore burn rate relative to cash reserves. Brilliant science is useless if the company runs out of money before completing trials.
Don’t fall for “story stocks” that articulate vision without demonstrating execution. In biotech, words are cheap. Clinical data is currency.
Don’t overreact to individual trial results. Clinical trials are experiments. A Phase 2 hiccup doesn’t invalidate the platform. Assess whether the strategic plan and budget account for potential delays.
Don’t invest in biotech without understanding the infrastructure. This isn’t an industry you can analyze through traditional valuation metrics. Understanding the machinery (R&D process, regulatory pathways, manufacturing challenges, partnership economics) is non-negotiable.
The XBI Insight: Structure Beats Selection
Here’s a fascinating structural observation: The SPDR S&P Biotech ETF (XBI) tracks the top biotechnology companies excluding big pharma.
In 1999, the top 100 public biotherapeutic companies had:
Smaller market cap than Merck alone
140 drugs in Phase 3 or awaiting FDA approval vs. Merck’s 5
65 drugs already on market vs. Merck’s 35
Even excluding pharma giants, biotech’s asset base (pipeline + products) exceeded individual pharma companies. The implication: biotech infrastructure had reached critical mass where collective innovation exceeded even the largest incumbents.
By 2025, this dynamic has intensified. The biotech sector contributed $396 billion in economic output to California alone, with over 17,200 establishments. Globally, bio-pharmacy holds a 42% market share, with biologics and biosimilars projected to grow at 13.2% CAGR through 2034.
The infrastructure doesn’t reside in any single company—it’s distributed across the ecosystem. Amgen handles manufacturing scale. Illumina provides sequencing. CRISPR Therapeutics offers editing platforms. Contract research organizations run trials. This modularity allows small companies to leverage ecosystem infrastructure without building everything in-house.
Part V: The Patent Cliff That Wasn’t — Why Copying Biology Is Harder Than It Looks
The $200 Billion Monopoly
On January 31, 2023, something remarkable happened in the U.S. pharmaceutical market: the world’s best-selling drug lost patent protection.
Humira—AbbVie’s monoclonal antibody treating rheumatoid arthritis, Crohn’s disease, and a half-dozen other autoimmune conditions—had generated over $200 billion in lifetime revenue. At its peak in 2022, it brought in $21 billion annually. That’s more than most biotech companies are worth, let alone what they earn in a year.
When a blockbuster drug loses patent protection, conventional wisdom says the bottom falls out. Generic competition floods in, prices plummet, and within months the branded drug becomes a footnote. This is what happened to Lipitor (Pfizer’s cholesterol drug) when it went generic in 2011. Within a year, generic atorvastatin captured over 90% of prescriptions.
So what happened when nine Humira biosimilars launched in the U.S. in 2023, some priced 85% lower than the $7,000-per-month branded product?
Humira retained over 80% of its patient base.
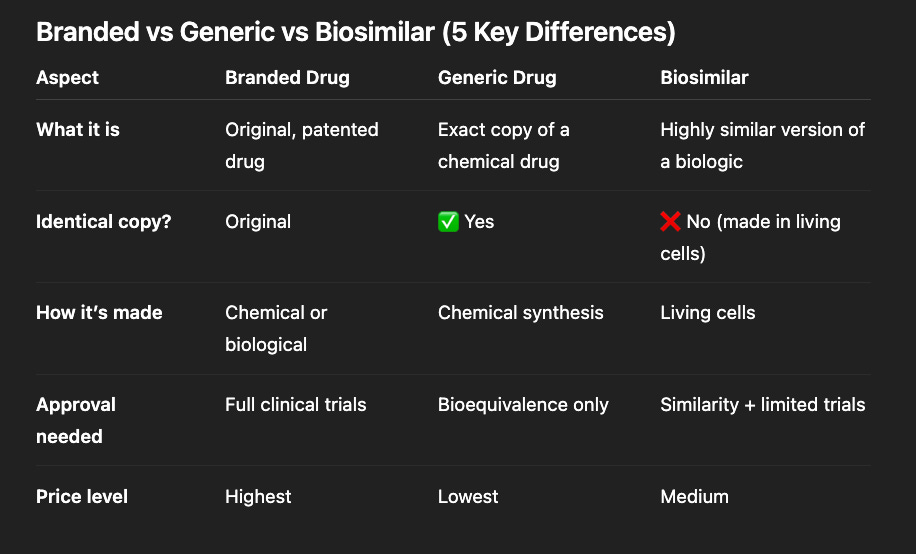
This wasn’t incompetence by biosimilar manufacturers. It wasn’t even primarily about AbbVie’s aggressive contracting tactics (though those helped). It was about a fundamental infrastructure reality: biosimilars are much harder to make than traditional generic drugs, resulting in fewer competitors.
The Two Patent Cliffs: Small Molecules vs. Biologics
To understand why biotech’s patent dynamics differ from traditional pharma, you need to grasp the manufacturing difference.
Small molecule drugs (like Lipitor, Viagra, or aspirin) are chemically synthesized. The molecular structure is relatively simple—usually a few hundred atoms arranged in a specific pattern. Once you know the structure, any competent chemist can reverse-engineer the synthesis pathway. Manufacturing involves standard chemical reactions in temperature-controlled vessels. Quality control means verifying the final molecular structure matches the original.
When Lipitor’s patent expired, within 12 months there were 10+ generic manufacturers producing atorvastatin. The market commoditized instantly.

Biologic drugs (mostly with DNA recombinant technology) are living organisms making proteins. The molecular structure is enormous—thousands of atoms folded into precise 3D shapes. You can’t simply synthesize a protein in a reactor. You must:
Insert the gene into living cells (bacteria, yeast, or mammalian cells)
Grow those cells in bioreactors (essentially massive fermentation tanks)
Harvest the proteins they produce
Purify extensively to remove cellular debris
Verify not just chemical structure but biological activity
Two proteins with identical amino acid sequences can have different biological effects if they fold differently or have different post-translational modifications (sugars attached, etc.). This is why they’re called “biosimilars” rather than “generics”—FDA-approved biosimilars must demonstrate they are as safe and effective as branded biologics through extensive testing, unlike traditional generics where automatic substitution is allowed.
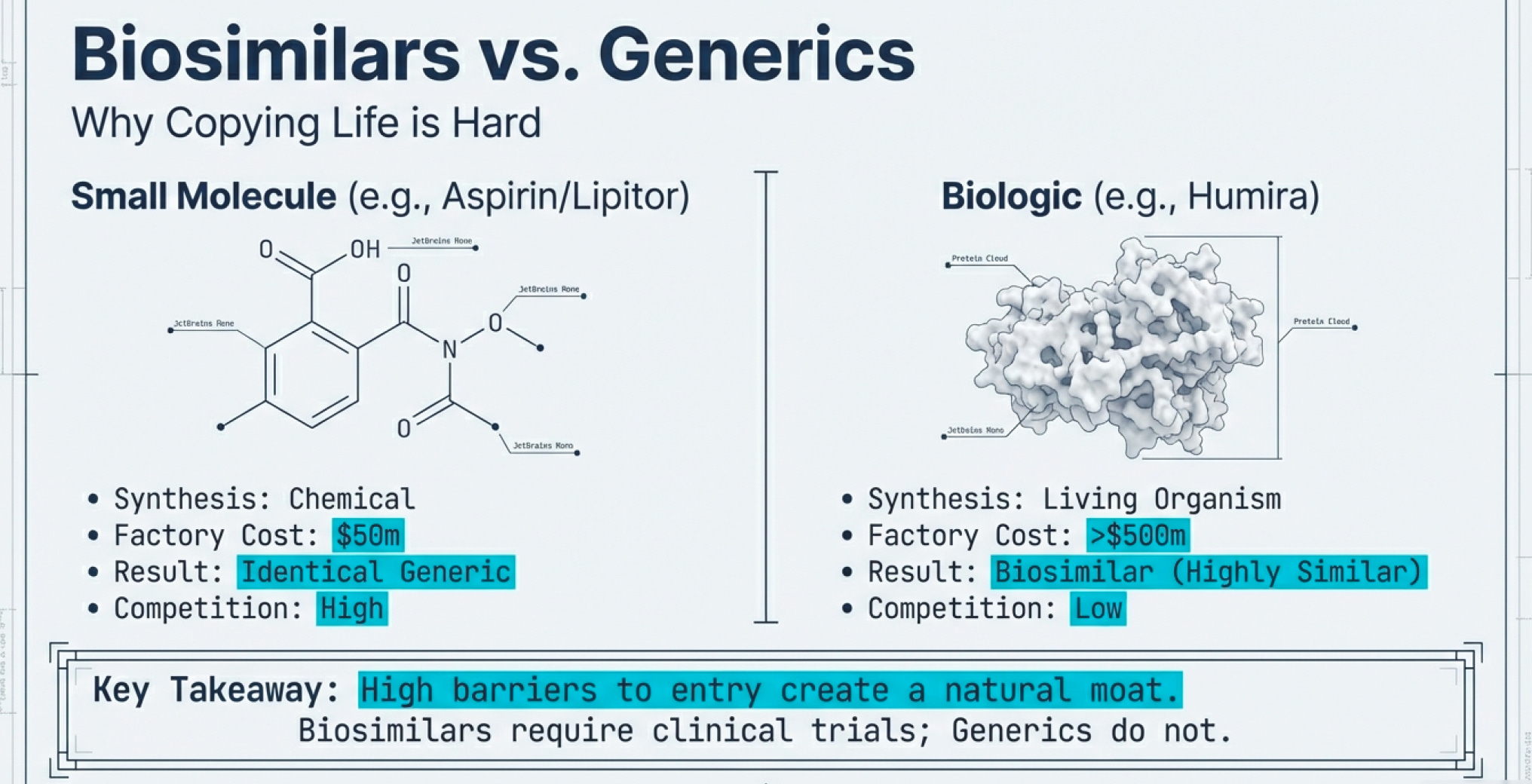

The infrastructure requirements are vastly different. A small molecule generic plant might cost $50-100 million to build. A biologics manufacturing facility? Try $500 million to $1 billion. And it takes years to validate that your process consistently produces the right protein with the right biological activity.
The AbbVie Master Class: How to Defend a Patent Cliff
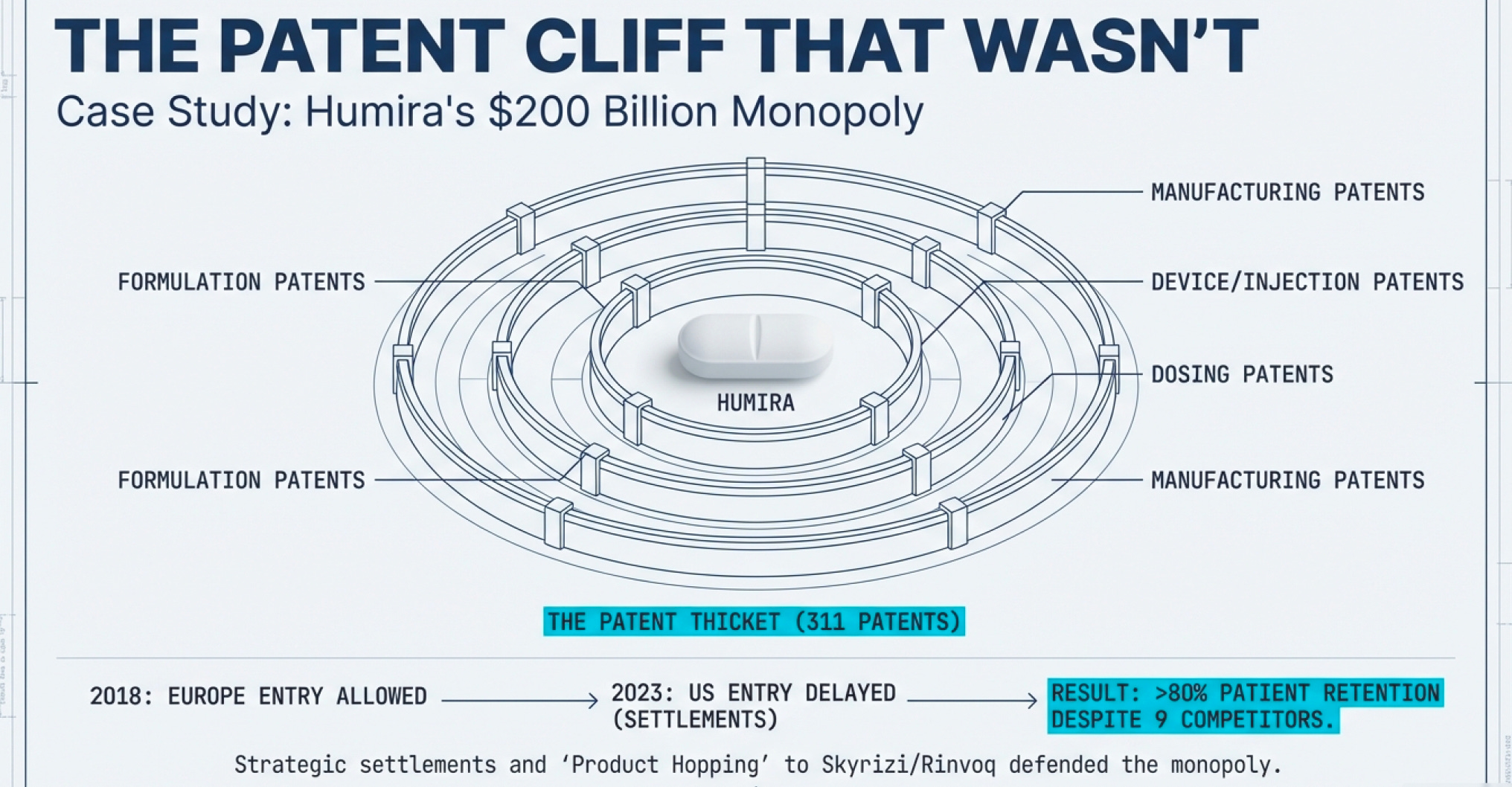
AbbVie’s Humira defense strategy deserves a case study in any business school (though perhaps not an ethics course). The company executed a three-part playbook that delayed U.S. biosimilar competition by five years and generated tens of billions in additional revenue.
Step 1: Build the Patent Thicket
AbbVie filed 311 patent applications for Humira in the U.S., with 90% filed after the drug received FDA approval in 2002. These weren’t all meaningful innovations. Many covered minor tweaks: the drug’s formulation, the injection device, the packaging, specific dosing regimens, manufacturing processes, even the citrate-free version (citrate caused stinging at the injection site).
Think of it like patenting a bicycle: the original patent covers the two-wheeled design. Then you file separate patents for the seat height, the handlebar angle, the gear shifter, the brake design, the wheel spoke pattern, and the paint color. Now anyone wanting to make a competitive bicycle must navigate dozens of patents, each of which could trigger expensive litigation.
According to a 2023 Matrix Global Advisors report, patent thickets on just five top-selling drugs cost the U.S. healthcare system over $16 billion in lost savings in a single year. The Humira delay alone—five years between European (2018) and U.S. (2023) biosimilar entry—cost the American healthcare system an estimated $14.4 billion to $19 billion.
Step 2: Strategic Settlement Agreements
Rather than fight every patent in court (which would take years and cost hundreds of millions), biosimilar manufacturers settled with AbbVie. The deals followed a pattern: AbbVie granted licenses to launch in Europe in October 2018, but secured commitments that biosimilar companies would delay U.S. market entry until 2023, and the biosimilar companies agreed to pay royalties to AbbVie on their future U.S. sales.
This is brilliant game theory. AbbVie effectively turned potential competitors into partners who had financial incentive (royalty payments) NOT to compete too aggressively. The settlements also staggered launches—Amgen launched first in January 2023, then seven more biosimilars in July—giving AbbVie time to adjust pricing and contracting strategies.
Step 3: Formulary Dominance and Next-Generation Products
While delaying biosimilar entry, AbbVie developed Skyrizi and Rinvoq—next-generation immunology drugs targeting the same conditions as Humira. When biosimilars finally launched, AbbVie had already started transitioning patients to these newer drugs, which had patent protection through the 2030s.
By Q4 2024, Skyrizi and Rinvoq combined sales were projected to surpass $31 billion by 2027, essentially replacing Humira revenue with new products. AbbVie’s European experience (where biosimilars launched in 2018) proved the playbook worked: despite list prices dropping up to 90% in some European countries, AbbVie retained market share through formulary dominance, tiered pricing, and expanded drug indications.
The result? Despite U.S. biosimilar launches in 2023 with discounts around 85%, Humira ceded only 2% of its market share to the five biosimilars that entered the market that year, according to Samsung Bioepis.
The Infrastructure Economics of Biosimilars
Here’s where the numbers get interesting—and reveal why biosimilar competition looks nothing like generic competition.
As of 2025, the FDA has approved 76 biosimilars, compared to over 30,000 approved generic drugs. Expensive biologic medications make up only 5% of prescriptions in the U.S. but account for 51% of total drug spending. Yet biosimilars account for only about 23% of the overall biologics market, with their market share remaining below 20% despite being approved as equally safe and effective.
Why the low penetration? Infrastructure constraints:
Manufacturing Complexity: Building biosimilar capacity requires specialized facilities. According to industry data, only a few biosimilar manufacturers will have attractive economics for each biologic molecule. The market structure more closely resembles complex generics like inhalation therapies, where three to five players typically enter in the first one to two years, rather than the 10+ competitors flooding oral solid dose generic markets.
Development Costs: While biosimilars require less R&D than originator biologics (since the target protein is known), development still costs hundreds of millions. Compare this to generic small molecules, where development might cost $1-5 million. The investment threshold creates a natural barrier to entry.
Regulatory Burden: The FDA in 2024 proposed guidance that biosimilars seeking interchangeable designation would no longer require switching studies, but biosimilars still face significantly more regulatory hurdles than traditional generics. Each biosimilar requires clinical trials demonstrating comparable efficacy and safety.
Switching Hesitancy: About 13% of patients who switch from Humira to an adalimumab biosimilar switch back to the originator. This “stickiness” reflects physician and patient comfort with existing treatments. Unlike generic pills (where pharmacists can automatically substitute), biologic switching often requires physician approval.
The Coming Tsunami: $200 Billion in Biologics Losing Exclusivity
Here’s what makes the next decade fascinating: over the next ten years, approximately $200 billion in revenues across 150 biologic molecules will face loss of exclusivity.
The casualty list reads like a “Who’s Who” of blockbuster drugs:
Keytruda (Merck): Merck’s cancer immunotherapy, now the world’s top-selling drug after dethroning Humira
Stelara (J&J): Treating psoriasis, psoriatic arthritis, and Crohn’s disease
Opdivo (Bristol Myers Squibb): Another cancer immunotherapy
Eylea (Regeneron): Treating macular degeneration
Xolair (Genentech/Novartis): Asthma and allergies
Competitive intensity will be particularly high for nine molecules whose expected annual brand revenue exceeds $5 billion, representing 45% of the total revenue pool.
The Biosimilar Market Growth Paradox
Despite all these challenges, the biosimilar market is experiencing explosive growth. The global biosimilars market is projected to grow from $34.75 billion in 2024 to $175.79 billion by 2034, at a 17.6% compound annual growth rate.
In the U.S., biosimilars generated $12.4 billion in savings in 2023, with total savings since their introduction reaching $36 billion, and more than 2.7 billion days of patient therapy with no meaningful differences in clinical outcomes.
The market is growing not because biosimilars are easy to make (they’re not), but because:
The prize is enormous: When biologics represent 51% of drug spending from just 5% of prescriptions, even modest market share gains generate billions in revenue
Payers are demanding alternatives: Insurance companies and government health programs are desperate to control costs
Infrastructure is maturing: More companies have built the specialized manufacturing capabilities required
Regulatory pathways are clarifying: FDA guidance continues evolving to reduce unnecessary testing requirements
North America led the biosimilars market in 2023 with 41% market share. But interestingly, Asia Pacific is expected to expand at the fastest CAGR of 18.5% during the forecast period, driven by lower manufacturing costs, growing patient populations, and government initiatives to reduce healthcare spending.
The Orphan Drug Exception: Where Patents Matter Less

Here’s a delightful paradox in biotech patent economics: sometimes the smallest markets have the best economics.
Orphan drugs treat rare diseases, defined as affecting fewer than 200,000 people in the United States or less than 5 in 10,000 people in the European Union. Big Pharma typically avoids these because fixed development costs can’t be recouped from small patient populations.
But for biotech companies, orphan drugs offer unique advantages:
Regulatory incentives: 7-year market exclusivity in the U.S., 10 years in Europe
Accelerated approval: FDA fast-track designation and priority review
Premium pricing: Limited patients means higher willingness-to-pay from payers
Lower competition: The small market naturally deters competitors
The result? Some of the highest-margin drugs in biotech treat rare diseases. BioMarin’s Vimizim (treating Morquio syndrome, affecting ~3,000 people globally) costs over $380,000 annually per patient. Alexion’s Soliris (treating paroxysmal nocturnal hemoglobinuria) costs over $500,000 per patient annually.
When your entire addressable market is 5,000 patients, you don’t worry much about biosimilar competition. The economics don’t work for Big Pharma/ competitors — the market is too small for them to put in large amount of effort and resource. It does not make sense for them.
This is why early-stage biotech companies often target rare diseases first: get an approved drug treating 10,000 patients at $300,000/year, and you have a $3 billion revenue stream—with almost no competition and strong patent protection.
Coda: The Unchanging Question
We’ve come full circle to 1999’s $6 billion question, now scaled 295x to a $1.77 trillion industry. The answer remains infrastructure: whoever controls the machinery for turning biological code into commercial products wins.
Recombinant DNA solved the protein extraction problem. Monoclonal antibodies solved the targeting problem. CRISPR is solving the genome editing problem. Each innovation becomes infrastructure—the foundation upon which thousands of companies build products.
The companies that matter aren’t necessarily those discovering new biology (though that helps). They’re the ones building innovative scalable systems for translating discoveries into therapies, diagnostics, and agricultural applications. Platform over product. Infrastructure over individual bets.
This is why small biotechs can compete with giants: infrastructure can be rented, licensed, or partnered. You don’t need Pfizer’s manufacturing network if you partner with Pfizer. You don’t need decades of regulatory experience if you hire experts who have it. You don’t even need your own clinical trial sites—contract research organizations handle that.
What you do need is a technology platform that solves a fundamental problem better than existing solutions. A way to turn code into cure more efficiently, more precisely, or more affordably.
The Cas9 enzyme editing DNA. The bacterial cell producing human insulin. The monoclonal antibody recognizing a tumor protein. These are the gears in biology’s invisible factory—and understanding those gears is the key to understanding biotech.
Not prediction. Infrastructure
Market data as of January 2025. Clinical trial information based on publicly disclosed results. This is educational content analyzing industry structure and competitive dynamics, not investment advice.
Adding an additional personal note in Biotech analysis: How to Analyze a Clinical Trial (Part 1 of 4 part series) - https://youtu.be/H_g8xjYufHo?si=PNJ6AybV01t1pZFL